

 上一版
上一版







 缩小
缩小 全文复制
全文复制 上一篇
上一篇
25岁的欣然喜欢大海,爱吃炸鸡,和许多女孩一样爱看《楚乔传》并害怕恐怖片。几年前她有过自己的事业:在淘宝上经营一家女包店,哪怕生意最好的时候,两三天才能卖出一个包。
曾经,她的生活跟其他同龄人并没太大差异。
可欣然身患大疱性表皮松解症(Epidermolysis Bullosa,简称“EB”) ,病情不断加重并出现各种并发症,她走路、吃饭、睡觉变得越来越费力,曾经的美好生活场景也渐渐成为记忆。
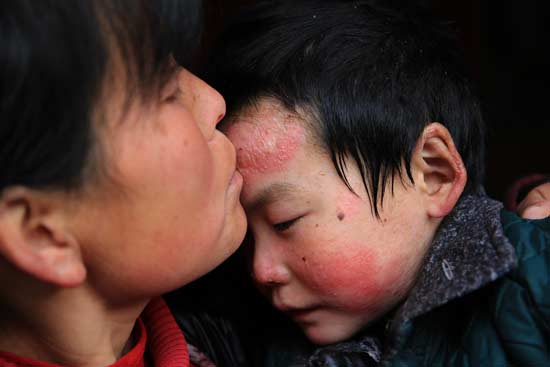
2017年10月底,在北京顺义举行的一场EB全国患者大会上,欣然成了诸多病友口中那个最令人心酸的女孩,看着眼前这个被纱布包裹全身布满伤口、手脚粘连挛缩成拳头,只能坐在轮椅上的女孩,人们忍不住叹气、摇头,又表情凝重地快速离开。
“这是一种由基因缺陷导致的单基因遗传性皮肤病,目前已经确定了19个致病基因。”北京大学第一医院皮肤科副主任医师林志淼介绍说,常染色体显性患者遗传给后代的概率是50%,常染色体隐性患者本身不会遗传给他的下一代,但患者的父母如果再要孩子,这个孩子有25%的概率会遗传。
EB患者的皮肤像蝴蝶翅膀一样极不结实,走路的压力、吃饭穿衣任何的摩擦都会导致身体的皮肤或黏膜反复起疱、破溃甚至脱落,随之而来的是剧烈难忍的疼痛及皮肤漫长的恢复期。他们被人善意地称作“蝴蝶宝贝”。
根据公益组织“蝴蝶宝贝”关爱中心的数据,目前我国登记在册的EB患者数量接近700位,多数为儿童。由于未经严格的流行病学调查统计,根据西方国家给出的1/20000~1/50000的患病概率估算,中国的EB患者有近万人。
与多数罕见病患者一样,现行医疗体制下巨大的城乡落差,导致“蝴蝶宝贝”面临专业诊疗人员不足、诊断不明确、易受歧视、药品可及性差、医疗保障机制不健全等问题。
治罕见病的医生比罕见病患者更罕见
1992年,欣然在山东聊城一家县医院出生时,右腿小腿处皮肤缺损一片,手指、脚趾也分布着大小不等的血疱,母亲王素琴当场吓蒙了。
由于从没见过这一类的患者,医生也查不出是什么病,凭着经验怀疑是“胎毒”或是孕妇怀孕时吃辣上火,只是对小欣然的起疱处或破溃处擦拭消毒水后用纱布进行简单包扎。
住院的20多天里经历了多次消毒、包扎,她的伤口终于结痂,王素琴长舒一口气抱着她出了院,可没过多久她就又起疱了。
在当地尝试了多种治疗方法无效后。这个平日里生活半径从未超出县城的母亲带着时满1岁的小欣然去了省会城市济南的山东省立医院,她在这里最终被确诊为重度营养不良型大疱性表皮松解症。
出生在沈阳的语彤是单纯型的EB患者,出生第九天就在一家大医院重症监护室里被确诊,但医生并没有给她提供有效的治疗方式,回家后家人发现孩子在床上蹬脚时脚起疱,穿尿不湿时肚脐也起疱,情况越来越不受控制。
语彤的母亲韩英抱着她去看了好多家医院,可医生都说这病不能治,有的三甲医院还把这病当成疱疹一类的皮肤病,开出各种药膏。
由于发病率低,患者基数小,受现行医疗水平所限,大部分EB患者都面临因诊断不明确、误诊继而辗转异地就医的处境,短则几个月,长则几年,错过最佳治疗期。
近15年来,国家儿童医学中心、北京儿童医院顺义妇儿医院皮肤科主任徐哲见过200位以上的EB患者,其中一半以上是在各地求医无果后经他人指点来北京求医的重型患者。让他特别难忘的是一个十来岁的西藏男孩,还没见面,诊室外就传来一股异样的气味儿,经验告诉他那是皮肤大面积破溃脱皮后血浆的味道。
男孩历经曲折找到徐哲时,身着大袍子,纱布粘在肉上,嘴唇周围有一块多年经久不愈的肉芽肿性皮损,手指挛缩,最终被确诊为重症交界型大疱性表皮松解症。
“当地老百姓传这孩子得什么怪病的都有,快10年了才确诊。”徐哲感慨男孩被耽误了这么长时间,他希望这种悲剧不再重演。
公益组织“罕见病发展中心”主任黄如方理解基层医院对EB确诊难的现实困境,北京、上海等一线城市的部分三甲医院,有条件对一些罕见病作出及时的诊断,但目前的医疗发展水平很难确保临床医生对每一种罕见病种都懂。
“(治疗)罕见病的医生比罕见病患者更罕见。”中日友好医院运动障碍与神经遗传病研究中心负责人顾卫红说。在近20年与患者打交道的过程中,她发现罕见病患者大部分的就医经历都非常曲折。她曾在多个公开场合展示过一张罕见病患者就医经历模式图,这幅图从导诊、专业医生就诊、基因检测、后续干预和注册随访等6个方面清晰地反映了罕见病患者的医疗路径。很多患者在获得明确诊断前都要走很漫长的路,有的人甚至因为求医心切,上当受骗,因病致贫。
顾卫红表示,现行医疗体系下巨大的地区差异使得罕见病专家集中于几个大城市,而患者则散居各地,很难获得精准的就医知识。此外,国内基因检测机构参差不齐的现状也间接导致患者难以获得明确诊断。
这是一种终生痛苦的疾病
多数罕见病患者群体因为行动不便或特殊的发病表现,很少会出现在公众的视野中,他们往往承受着常人难以想象的病痛折磨和社会歧视。
欣然到了上学前班的年龄时,被母亲王素琴领着去学校。负责的老师只是瞧了一眼孩子,没多考虑就拒绝了。校长回复王素琴说:“像您这孩子,要是其他孩子碰了她,我们也有责任;您说这病不传染,可其他家长有意见来找我,我没法解释啊。”
之后的二十几年里,欣然再也没能去上学,只能靠退休在家的爷爷教一些简单的知识。
求学被拒,求医路上也要忍受路人投来的异样眼光。欣然每次出门,总有人在背后指点议论:“瞧,这孩子被烧成了这样。”有些父母一看见她的模样就一把拉开自己的孩子,躲得远远的。
不仅如此,EB患者的普通就医需求也常常得不到满足,一些患者看牙科,或者去看消化科,常常会因为伤口伤情的特殊性被拒诊,有时连抽血化验都有困难。
2012年,周迎春发起了国内首个“蝴蝶宝贝”关爱中心,致力于为患者提供政策支持,对接医疗资源,提供医疗帮助。据他了解,一些患儿甚至连打疫苗都会遭拒。碰到这种情况,他会让家长去几家三甲医院的皮肤科开具可以接种疫苗的证明,但也有的防疫站以防疫体系和医疗体系是两条线为由拒绝承认证明的有效性。
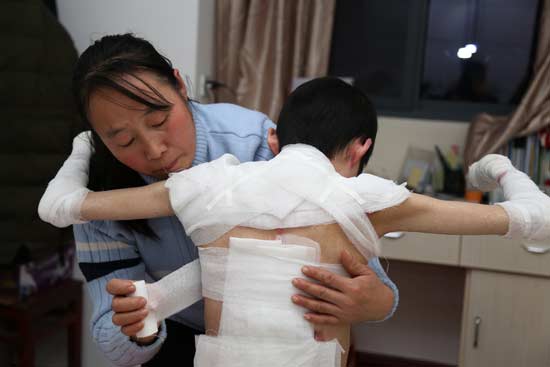
除了误解带来的歧视问题,EB患者的生活质量也很低。徐哲表示,目前该疾病没有有效的治疗方法,多数只能通过美皮康、优拓等敷料来进行包扎护理,且护理费时费力。按照患者皮肤受伤的严重程度估算,戳水疱、清创、更换敷料、包扎,一个完整的流程下来,需要耗费2~4个小时不等。多数家庭中,父母至少有一方辞职在家照看孩子。
“蝴蝶宝贝”关爱中心理事长周迎春也是一位EB患者的父亲,2003年8月,女儿周敏出生,那之前他担任上海一家通讯公司的部门经理,月薪1万多元。女儿出生后,每天检查新水疱、清创、更换敷料、包扎伤口一整套流程下来接近4个小时。为了腾出时间给孩子做护理,先是同在一家公司工作的妻子辞了工作,之后他也从经理变成了一名普通职员。
“这是一种终生痛苦的疾病。”林志淼医生说,日常生活的每一个动作都有可能给他们带来剧烈疼痛。比如衣服的标签如果不及时拆除,就可能会摩擦导致患者颈部出现水疱。
如果一定要有一个治疗方案的话,骨髓移植是一个选择,但费用高、风险大,治愈效果依旧有限,并不是一劳永逸的方式。骨髓移植目前只适用于严重泛发型营养不良型等重型的患者。“国际上接受过骨髓移植的患者有二十几例,有1/3效果明显,1/3效果不明显,剩下1/3死亡。”林志淼强调,哪怕是移植成功的患者也具有不确定性,他们可能不断地同感染和排异作斗争。
周敏是国内EB患者中接受骨髓移植的第一人,在病情恶化到有一半时间食道疼得喝不下水的情况下,她决定赌一把。从2014年做完移植存活的3年多来,她分别经历了肺炎、脑膜炎、自身免疫性溶血3次大的感染,进过一次重症监护室。
非药物和手术治疗的关键在于合理保护伤口,减少摩擦,正确护理。徐哲强调:“如何护理非常关键,如果患者能在医疗上得到良好的帮助,尽早进行正确护理,就能够大大延缓他的痛苦,降低死亡率。”
而专科医务人员的欠缺恰恰是目前最大的问题,能开展正确护理的医生和护理师更是罕见。林志淼坦言,在专业医生的指导下依靠患者组织来给患者家属培训,传授护理经验成了目前的权宜之策。
据徐哲介绍,目前分别设在首都医科大学附属北京儿童医院、北京儿童医院顺义妇儿医院和北京大学第一医院这3家医院的国内首批“蝴蝶宝贝”专病门诊正在进行积极探索,帮助患者及时诊断,并提供几十年预后的专业诊疗护理指导,从孩子出生到上学以及工作,都能提供不同的护理方法。具体包括指导使用不同的敷料、不同季节的穿衣原则、不同的伤口处理方式等。
如何走出罕见病医疗困境
护理之外,获得廉价好用的外用药是EB诊疗的最终追求。罕见病用药也被称为“孤儿药”, “孤儿药”既贵且缺是罕见病群体面临的最大医疗困境。
我国目前并没有明确的罕见病定义。2010年5月,中华医学会遗传学分会在上海组织召开了中国罕见病定义专家研讨会,结果建议:患病率低于1/500000,或新生儿发病率低于1/10000的疾病,可以称为中国的罕见病。
事实上,由于我国人口基数大,“罕见病”并不罕见。2017年9月8日,北京协和医院副院长张抒扬在第六届中国罕见病高峰论坛上表示:按国际方法计算,中国有超过3000万罕见病患者。
就EB患者而言,目前大多敷料都依赖进口产品,按照市场价,一片10厘米见方的“美皮康”售价30多元,“优拓”也得10多元,患者自行购买每月光敷料就得花上千元。
周迎春给记者算了一笔账。在周敏接受骨髓移植前,他每年光买敷料就得花五六万元。然而现行的医保政策并没有细化到具体罕见病的用药,敷料大多不能通过医保报销,为了节约经费,很多患病家庭会将一次性的敷料洗干净后重复使用。
王素琴常常感叹日子艰难,为了维持女儿的身体状态,她每月都得花上千元来购买敷料、纱布和红霉素眼膏等药物,这还不包括女儿发生各种并发症住院的花销。每次临近买药前的几天,她的眉头都难以舒展,丈夫辞职在家,她一个月的工资扣除养老保险剩下不到1300元。
如今靠着一些企业的捐赠,“蝴蝶宝贝”关爱中心向登记在册的患者免费发放部分敷料,在一定程度上减轻了患者的经济负担。
目前全球已知的约7000种罕见病病种当中有80%是遗传性疾病,近50%的受累人群是儿童,受影响人数超过3亿。而数据显示,这其中仅5%的罕见病有药可医,黄如方强调,在这5%当中只有30%的药已在中国上市,且大多昂贵,多数患者难以承受。
顾卫红进一步指出,由于患者基数小、市场容量小, “孤儿药”回报率较低,这限制了药企对于此类药品的研发力度。
我国“孤儿药”的自主研发上市更是不容乐观。“最主要还是国家的重视程度问题,目前我国并没有特别好的罕见病药物研发的激励措施来引导医疗企业去投入研发。”黄如方说。
美国早在1983年就通过了有关法案,对参与“孤儿药”研发的公司提供快速审批、财政支持药品临床试验费用的50%、给予研发生产企业7年市场独占期等在内的多项支持。法案通过至今,有近500种“孤儿药”获准上市,而在法案出台之前的10年内,仅有10种获批。此外,日本对罕见病用药研发公司给予10%的税务减免,韩国对罕见病用药报销2/3的费用,欧盟则给予研发企业长达10年的市场独占期。
有了药但药价高患者吃不起,还必须解决一个由谁支付的问题。我国的社会医疗保险带有“低水平、广覆盖”的性质,医保基金的承受能力不高。
“一些‘孤儿药’一年的药物费用就得上百万元,如果将这些都纳入医保,医保的来源在哪里?”顾卫红指出,国内有识之士一直在探索的解决路径,是在基础医疗保障体系的基础上确立医保、商业保险、慈善组织、患者个人多方负担的筹资机制,以降低罕见病治疗费用的自付比例。
尴尬的现实是,我国至今没有出台官方的罕见病目录,绝大多数罕见病用药没有纳入医保目录。黄如方建议,政策制定者应首先将有药可用且患者数量相对多的几类罕见病用药纳入医保目录。
而解开这些难题的关键就在于制定中国的《罕见疾病防治法》。
顾卫红认为,现阶段我国罕见疾病立法至关重要,但是目前罕见病种类有7000多种,这些疾病在病因和发病机制研究、诊断和治疗方面处于不同的阶段,患病人数也各不相同。罕见病问题本身也早已超越了单纯的医学问题,如何开始立法工作成了一个难点。“必须开始才能逐步完善,目前出台第一批罕见病名录仍十分必要,探索积累经验后可陆续修订名录。”顾卫红说。
黄如方也认为,现阶段我国罕见病立法的方向是首先要明确罕见病定义,出台明确的激励措施以鼓励药企投入研发。
我国已出台多项措施尝试为“孤儿药”的研发和及早上市打开通道。2017年10月,中共中央办公厅、国务院办公厅印发《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》中,明确了支持罕见病治疗药品医疗器械研发,公布罕见病目录,建立罕见病患者登记制度,罕见病治疗药品医疗器械注册申请人可提出减免临床试验的申请,对罕见病治疗药品给予一定的数据保护期等内容。
此外令人欣慰的是,2017年12月20日,国家食品药品监督管理总局对外公布了《拓展性同情使用临床试验用药物管理办法(征求意见稿)》,允许在开展临床试验的机构内使用尚未得到批准上市的药物给急需的患者。这被称作中国版的“同情给药制度”。有评论指出,“同情给药制度”的目的,就是为了跳过药品审批的繁缛过程,以最简捷的路径为患者提供目前唯一可能有效的治疗。
“立法之后要有实施细则才行。罕见疾病的立法或许正在解冻,但实施起来肯定不容易,而这需要全社会包括医疗界、公益领域、政府的相关职能部门、学术界等多方共同努力才能实现破冰。”顾卫红说。
(文中欣然、王素琴、语彤、韩英、周敏均为化名)




