

 上一版
上一版





 缩小
缩小 全文复制
全文复制 上一篇
上一篇
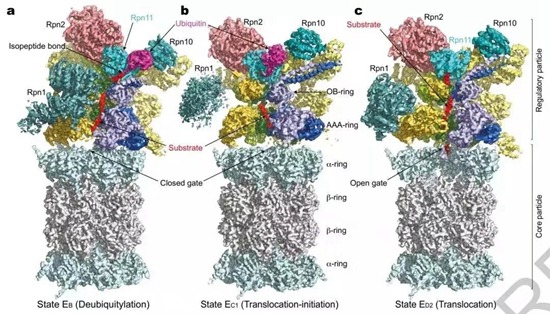
11月12日,北京大学物理学院人工微结构和介观物理国家重点实验室、前沿交叉学院定量生物学中心毛有东课题组在《自然》(Nature)杂志在线发表了题为Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome(《底物结合的人源26S蛋白酶体的冷冻电镜结构和动力学》)的研究论文。据介绍,这是《自然》上发表的系统性的、优于3.6埃分辨率水平实验研究超大复合蛋白质机器的动力学过程和原理的第一篇论文,标志着冷冻电镜的发展终于开始进入期待已久的全原子动力学分析的新时代,被审稿人誉为相关领域的“里程碑”。
据悉,由于《自然》编辑部和审稿人对该论文发表极其重视,该论文从今年9月11日投稿、审稿到11月12日正式Online,仅用两个月时间,以加速出版的形式在线发表。
泛素-蛋白酶体体系(Ubiquitin-Proteasome System,简称UPS)是细胞内最重要的蛋白质降解通路,对维持生物体内蛋白质的浓度平衡,以及对调控蛋白、错误折叠或受到损伤的蛋白的快速降解起着至关重要的作用,参与了细胞周期、基因表达调控等多种细胞进程,由UPS失常引发的蛋白质新陈代谢异常与众多人类重大疾病直接相关。
尽管上述工作揭示了蛋白酶体的基本架构和内在运动行为,但由于缺乏蛋白酶体与底物之间的相互作用,人们对于蛋白酶体如何实现底物降解的原子水平工作机制仍一无所知。此外,尽管冷冻电镜技术近年来广泛应用于分析具有动态特征的蛋白复合体结构和平衡态构象,但对其中间态结构和非平衡构象分析的分辨率水平往往局限在4~6埃或更低,离真正的全原子水平动力学分析还有相当一段距离。
而该论文通过冷冻电子显微镜和机器学习技术的结合,解析了人源蛋白酶体26S在降解底物过程中的7种中间态构象的高分辨(2.8~3.6埃)精细原子结构,最好局部分辨率达2.5埃。
据北京大学介绍,这些研究结果为几十年来对蛋白酶体功能的研究提供了宝贵的第一手原子结构和动力学信息,对于理解生物体内蛋白质的降解过程和一系列负责物质输运的ATPase马达分子的一般工作原理具有极为重要的科学意义,也标志着北大冷冻电镜平台的建设后来居上,在数据采集效率和成像分辨率等各方面已达到国际领先水平。
中国青年报·中青在线记者 叶雨婷




